

Electronic Structure Book :: Errata
For a list of most important errata (up to May 2005)
Individual Errata by page number:
Back Cover Pages: xxi, 2, 70, 72, 75, 83, 84, 97, 127, 252, 261, 287, 296, 472, 479-80, 575
Information organized by Table of Contents
- Cover
- Contents, Preface, Notation
- Part I: Overview and background topics
- Ch. 1. Introduction
- Ch. 2. Overview
- Ch. 3. Theoretical Background
- Ch. 4. Periodic solids and electron bands
- Ch. 5. Uniform Electron Gas and Simple Metals
- Part II: Density Functional Theory
- Ch. 6. Density functional theory: Foundations
- Ch. 7. The Kohn-Sham Ansatz
- Ch. 8. Functionals for exchange and correlation
- Ch. 9. Solving the Kohn-Sham equations
- Part III: Important Preliminaries on Atoms
- Ch. 10. Electronic Structure of Atoms
- Ch. 11. Pseudopotentials
- Part IV: Determination of Electronic Structure: The Three Basic Methods
- Ch. 12. Plane Waves and Grids: Basics
- Ch. 13. Plane Wave methods: Full Calculations
- Ch. 14. Localized Orbitals: Tight Binding
- Ch. 15. Localized Orbitals: Full Calculations
- Ch. 16. Augmented Functions: APW, KKR, MTO
- Ch. 17. Augmented Functions: Linear Methods
- Part V: Predicting Properties of Matter from Electronic Structure - Recent Developments
- Ch. 18. Quantum Molecular Dynamics (QMD)
- Ch. 19. Response Functions: Phonons, Magnons, ...
- Ch. 20. Excitations Spectra and Optical Properties
- Ch. 21. Wannier Functions
- Ch. 22. Polarization, localization and Berry's phases
- Ch. 23. Locality and linear scaling O(N) methods
- Ch. 24. Where to find more
- Appendices
- Color Versions of Figures in Text
- Contact the author
Cover
Erratum - Back Cover:
The author has finished his term as associate editor of Reviews of Modern Physics; Peter Littlewood is now associate editor for condensed matter theory.
Contents, Preface, Notation
Erratum - Page xxi, Notation - u alpha beta denotes a strain tensor (not a stress tensor)
Ch. 1. Introduction
Erratum - Page 2, line 6 - 1991 should be 1911
Ch. 2. Overview
Color Figures:
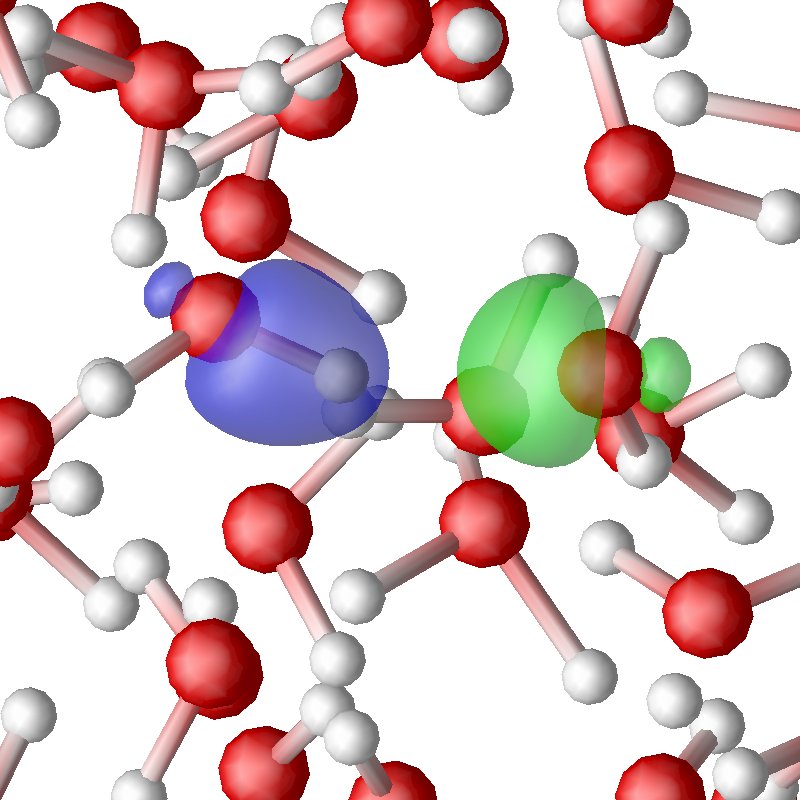
Figure 2.12. Snapshots of simulation of water. Figure 2.12a., Figure 2.12a., Figure 2.12a.,provided by E. Schwegler
Nanotubes: many sites for structures and properties of nanotubes can be found in the nanotube folder. See other useful sites. (Note that there are different definitions. The site "Nano-tube & Nano-cone Applet by (c)S.Weber" constructs nanotubes denoted by the number of atoms around the tube, whereas the standard notation for nanotubes used in the book is in terms of the repeat vector in units of the graphene vectors. Thus a (13,0) nanotube contains 12 units in the circumference and is termed (12,0) in "Nano-tube & Nano-cone Applet by (c)S.Weber".)
{kind=link}
{kind=link}
{kind=link}
Ch. 3. Theoretical Background
Erratum - Page 70: revised text
Erratum - Page 72:
Exercise 3.19 is incorrect as stated. The second sentence should read: Show that such an empty orbital does not experience a self contribution to the exchange energy, whereas for a filled state there is an attractive self term in the exchange.
Ch. 4. Periodic solids and electron bands
Erratum - Page 75: Caption of Fig. 2 - "60 degrees" should be replaced by "90 degrees" in two places.
Erratum - Page 83: Eq. (4.14) - for fcc, b3 should be (-1,1,1).
Erratum - Page 84: Eqs. (4.16) and (4.17) contain spurious "|;". These should be omitted.
Erratum - Page 97: Exercise 4.3 - "60 degrees" should be replaced by "90 degrees". Crystal Lattice Structures Web site with figures and information on crystal structures designated by any of the conventions: Strukturbericht Designation, Pearson Symbol, or Space Group. Also Structures of Intermetallic Alloy Phases Bilbao Crystallographic Server This site provides extensive information on crystals classified in terms of symmetry: Figures of the Brillouin Zone with labelled high symmetry lines and points for any Space Group, Generators and General Positions of Space Groups, Wyckoff Positions of Space Groups Nanotubes: many sites for structures of nanotubes can be found in the nanotube folder. See other useful sites. (See note in Chapter 2 on differences in definitions of the notation for tubes.)
Ch. 5. Uniform Electron Gas and Simple Metals
Ch. 6. Density functional theory: Foundations
Erratum - Page 127: Errors in Eqs. (6.20) - (6.21). revised text
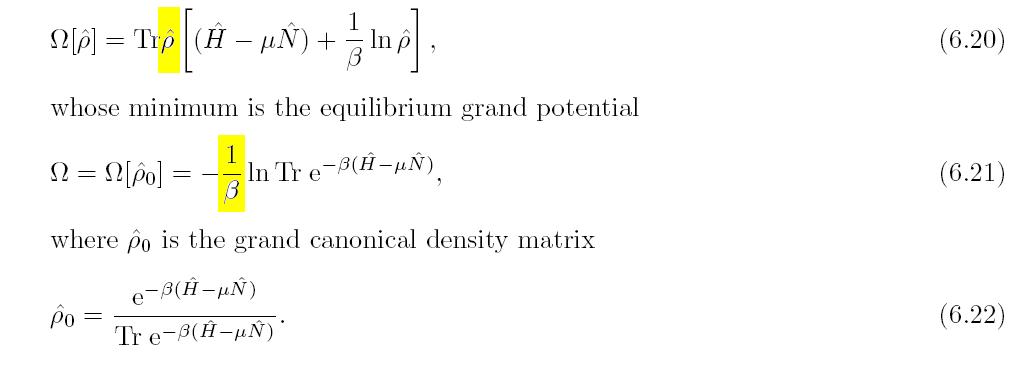{kind=link}
Ch. 7. The Kohn-Sham Ansatz
Color Figures: XC hole in Si related to Figure 07-03 provided by R. Q. Hood
Ch. 8. Functionals for exchange and correlation
Ch. 9. Solving the Kohn-Sham equations
Ch. 10. Electronic Structure of Atoms
- NIST Atomic Reference Data for Electronic Structure Calculations
- NIST Basic Reference Data for Electronic Structure Calculations
- A multi-configuration Hartree-Fock program by Charlotte Froese Fischer which is described in Computer Physics Communications Volume 1, Issue 3 , January 1970, Pages 151-166.
- The Herman-Skillman code is a classic Hartree-Fock code dsecribed in the book: Herman, Frank and Skillman, Sherwood, Atomic Structure Calculations, Prentice-Hall, Inc., Englewood Cliffs, New Jersey, 1963. It is one of the simplest Self-Consistent Field computational codes for determining one-electron wave functions and the associated potential for any free atom or ion.This site contains versions from "quasi-original" to an updated F77 form.
Chapter 11 - Pseudopotentials
See also information in links in Planewave methods II
- Pseudo-Dojo. The PseudoDojo.org website gives access to the latest released version of the pseudopotentials of the PseudoDojo project. The PseudoDojo project is hosted on github. It is a python package to develop, test and store pseudo potentials. The formats are: psp8, upf, psml, xml, html, djrepo.
- Pseudopotential programs maintained by José Luìs Martins. Consruction of Troullier-Martins, Hamann-Schluter-Chiang, and Kerker pseudopotentials, including semi-relativistic effects and several flavours of the density functionals.
- The Fritz-Haber Institut fhi98PP pseudo potential program. The package fhi98PP [1] allows one to generate norm-conserving pseudopotentials of the Hamann Troullier-Martins types, employing common parameterizations of the LDA and GGA functionals. Also one can check the transferability of the pseudopotentials and search for unphysical ghost states [in the separable (Kleinman-Bylander) representation. A hands-on tutorial and worked-out examples are available. [1] M. Fuchs, M. Scheffler, Comput. Phys. Commun., Comput. Phys. Commun. 119, 67-98 (1999), "Ab initio pseudopotentials for electronic structure calculations of poly-atomic systems using density-functional theory"
[pdf (reprint)], [abs, src, ps], European mirror: [abs, src, ps] - Ultrasoft Pseudopotential Generation Code and Library. This is a publicly available software package for generating ultrasoft pseudopotentials. It also includes a library of available pseudopotentials for some atoms of the periodic table.
Chapter 12 - Plane Waves and Grids: Basics
- Erratum - Page 252, line 2: Mathieu is the correct name (not Matthew) Page 252, line 4: Exercise 12.4 (not 12.7).
- TBPW is an electronic structure code for tight binding (TB) and plane wave (PW) calculations. It is primarily intended for pedagogical purposes; written from the ground-up in a modular style using Fortran 90 with parts common to all band structure codes in modules in a common directory. Various TB models and empirical pseudopotentials are included and it is constructed so that users can add others. Bands are produced in a form displayed by gnuplot. The TB models include simple cases specified by simple inputs or a fully general Slater-Koster 2-center form for arbitrary angular momenta as described in Appendix N.
- Exercise 12.4 - Added note: The differential equation with the cosine potential was solved by the French mathematician Emile Léonard Mathieu (born 1835 in Metz, died: 19 Oct 1890 in Nancy, France). The equation is known as the Mathieu equation and the solutions, as the Mathieu functions. Solutions in one, two and three dimensions are considered in a paper by J. C. Slater, Phys Rev 87, 807 (1952). (Thanks to Oswaldo Dieguez.)
Chapter 13 - Plane Waves II: self-consistent density functional calculations
Erratum - Page 261, bottom: Reference [560] should be [567]
- ABINIT ABINIT is an Open Source Free Software code (GNU license) for electronic structure calculations using norm-conserving pseudopotentials and plane waves. It allows relaxation of the atoms and MD simulations and it includes response functions. Excitations can be calculated using TDDFT and GW many-body methods.
- PARATEC Home Page PARAllel Total Energy Code - PARATEC is a package designed primarily for a massively parallel computing platform and can run on serial machines. The code performs total energy calculations using a plane wave basis and norm-conserving pseudopotentials (typically Hamann-Schulter-Chiang or Trouillier-Martins). Forces and stress can be calculated and used to relax the atoms into their equilibrium positions.
- QE Quantum Espresso is a set of programs for electronic structure calculations within Density-Functional Theory and Density-Functional Perturbation Theory, using a Plane-Wave basis set and pseudopotentials. QE is released under the GNU General Public License.
- VASP (also (VAMP) Vienna Ab Intio Simulation Package VAMP/VASP is a package for performing ab-initio quantum-mechanical molecular dynamics (MD) using pseudopotentials and a plane wave basis set. The approach is based on a finite-temperature method and evaluation of the instantaneous electronic ground state at each MD-step using efficient matrix diagonalization schemes and Pulay mixing. The interaction between ions and electrons is described using ultrasoft Vanderbilt pseudopotentials (US-PP) or the projector augmented wave method (PAW), especially useful for transition metals and first row elements. VAMP/VASP is not public domain - if you are interested, contact Juergen.Hafner@univie.ac.at
Chapter 14. Tight binding
Erratum - Page 287: In Eqs. (14.15) and (14.16) the x and y components should be interchanged to agree with the cell oriented as in Fig. 4.5 and 14.9a. The K point given below (14.16) should be (kx = (2/3)(2 pi/a), ky = 0). Erratum - Page 296, Exercise 14.19: The K point should be (kx = (2/3)(2 pi/a), ky = 0).
- OHMMS - Object-oriented High-performance Multiscale Material Simulator. Simulations using many methods for molecular dynamics and long time scales. Forces are derived from empiricial potentials or tight-binding electronic structure methods. Work in progress is to couple to full DFT calculations.
- TBPW is an electronic structure code for tight binding (TB) and plane wave (PW) calculations. It is primarily intended for pedagogical purposes; written from the ground-up in a modular style using Fortran 90 with parts common to all band structure codes in modules in a common directory. Various TB models and empirical pseudopotentials are included and it is constructed so that users can add others. Bands are produced in a form displayed by gnuplot. The TB models include simple cases specified by simple inputs or a fully general Slater-Koster 2-center form for arbitrary angular momenta as described in Appendix N.
- Nanotubes illustrate the tight-binding method. Many sites for structures of nanotubes can be found in the nanotube folder. See other useful sites. (See note in Chapter 2 on differences in definitions of the notation for tubes.)
Chapter 15. Local-orbital methods
- Siesta (Spanish Initiative for Electronic Simulations with Thousands of Atoms) uses pseudopotentials and numerical local orbitals to perform electronic structure calculations and ab initio molecular dynamics simulations of molecules and solids.
Ch. 16. Augmented Functions: APW, KKR, MTO
Ch. 17. Augmented Functions: Linear Methods
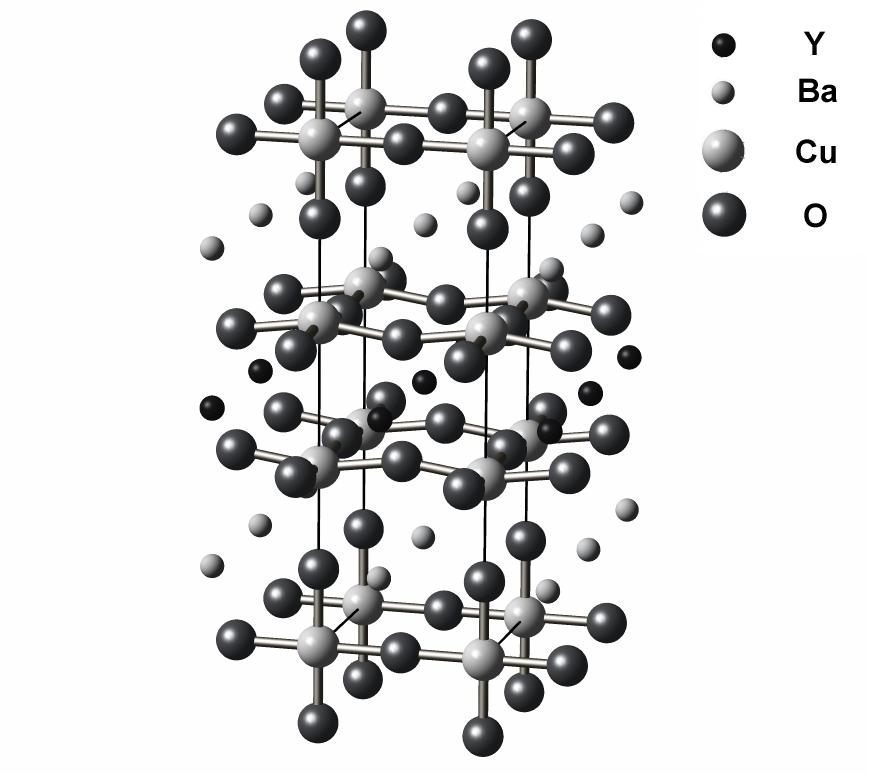
Figures: Figure 17-03 provided by W. E. Pickett, Figure 17-10 provided by O. K. Andersen.
{kind=link}
Ch. 18. Quantum Molecular Dynamics (QMD)
Ch. 19. Response Functions: Phonons, Magnons, ...
Ch. 20. Excitations Spectra and Optical Properties
- Octopus - time-dependent (TDDFT). Excitation spectra are computed using time-dependent form (TDDFT) by performing simulations in time. is based on a nuclear-physics code written by Bertsch and Yabana for real-time dynamics using real space grid an finite difference methods.
Ch. 21. Wannier Functions
- Webpage for Maximally-localized Wannier functions. This site provides references and a publically available software package for postprocessing a set of Bloch functions to obtain a maximally localized set of Wannier functions.
Ch. 22. Polarization, localization and Berry's phases
Ch. 23. Locality and linear scaling O(N) methods
Erratum - Page 472, last sentence of the last paragraph of section 23.7: The correct reference [859] for localized basis sets that are overlapping spherical waves should be: P. D. Haynes and M. C. Payne, "Localised spherical-wave basis set for O(N) total-energy pseudopotential calculations", Comput. Phys. Commun. 102, pages 17-27 (1997). The reference given describes a trsansfornation of plane waves to "Non-orthogonal Generalised Wannier Functions" which are optimised directly. The linear-scaling implementation is called ONETEP and is described in links at http://www.onetep.org/
Ch. 24. Where to find more
Appendices
Appendix A. Functional equations Appendix B. LSDA and GGA functionals. Erratum - page 479-80: Eqs. (B.4) and (B.5) should be omitted; they repeat (B.2) and (B.3) and contain small errors. In Eq. (B.6) the letters "m" and "M" are spurious and should be removed. In the first line of (B.6) n(rs) should be ln(rs) in both places. Note that only selected forms for the unpolarized case are given; complete expressions can be found in [224,368,413]. Appendix C. Adiabatic approximations Appendix D. Response functions and Green’s functions Appendix E. Dielectric functions and optical properties Appendix F. Coulomb interactions in extended systems Appendix G. Stress from electronic structure Appendix H. Energy and stress densities Appendix I. Alternative force expressions Appendix J. Scattering and phase shifts Appendix K. Useful relations and formulas Appendix L. Numerical methods Appendix M. Iterative methods in electronic structure Appendix N. Code for empirical pseudopotential and tight-binding: schematic description
- TBPW is an electronic structure code for tight binding (TB) and plane wave (PW) calculations. It is primarily intended for pedagogical purposes; written from the ground-up in a modular style using Fortran 90 with parts common to all band structure codes in modules in a common directory. Various TB models and empirical pseudopotentials are included and it is constructed so that users can add others. Bands are produced in a form displayed by gnuplot. The TB models include simple cases specified by simple inputs or a fully general Slater-Koster 2-center form for arbitrary angular momenta as described in Appendix N.
Appendix O. Units and conversion factors. Erratum - page 575: The speed of light in atomic units should be 137.036,000. The NIST tabulation of latest recommended values fundamental physical constants can be found at: http://physics.nist.gov/cuu/Constants/
Color Versions of Figures in Text
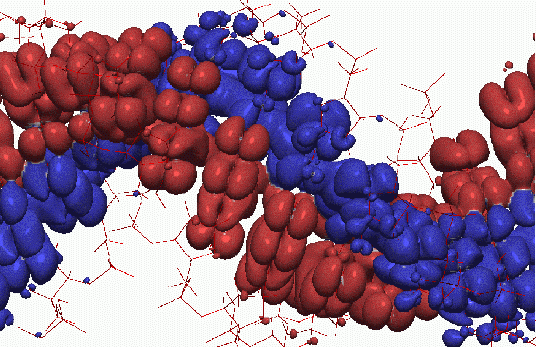
Figure 23.9. Wannier function for amorphous Si (1.8 Mbytes), zipped version (171 Kbytes) created by U. Stephan. Figure 23.10. Eigenstates of DNA molecule provided by E. Artacho
{kind=link}